
Les actualités


Les projets à la une
Découvrez la Fondation pour l'Innovation en Cardiométabolisme et Nutrition
Situé au coeur de l’hôpital de la Pitié-Salpêtrière à Paris, l’ICAN est un pôle d’excellence dans le domaine des maladies du cardiométabolisme : diabète, obésité, maladies hépatiques (stéatose), maladies du cœur et des vaisseaux.
168Médecins
221Chercheurs
186Articles de consensus et recommandations internationales
4Parcours de soins innovants
6Centres de référence Maladies rares
5167Publications scientifiques


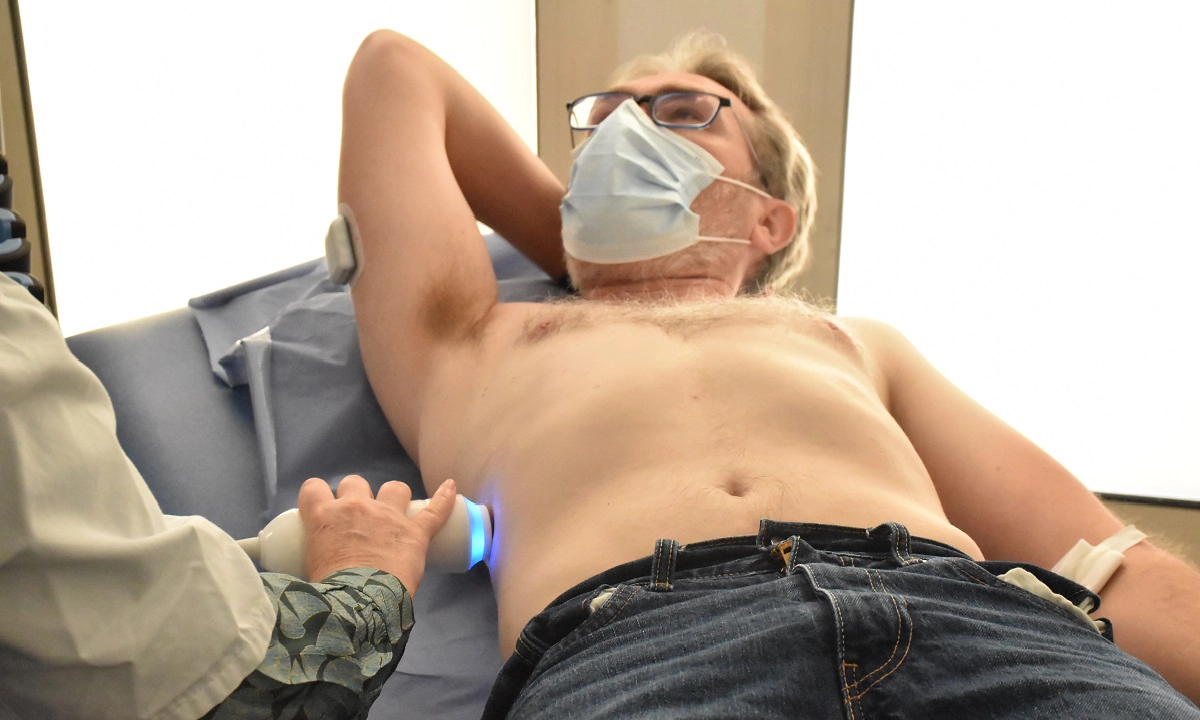
Soigner les maladies cardiométaboliques par la recherche translationnelle
Nous rassemblons toutes les expertises scientifiques et médicales pour accompagner les patients tout au long de leur parcours de vie : prévention, diagnostic précoce, prise en charge globale et personnalisée, épisodes aigus, suivi des maladies chroniques
Diabète
Obésité
Coeur et vaisseaux
Stéatose du foie

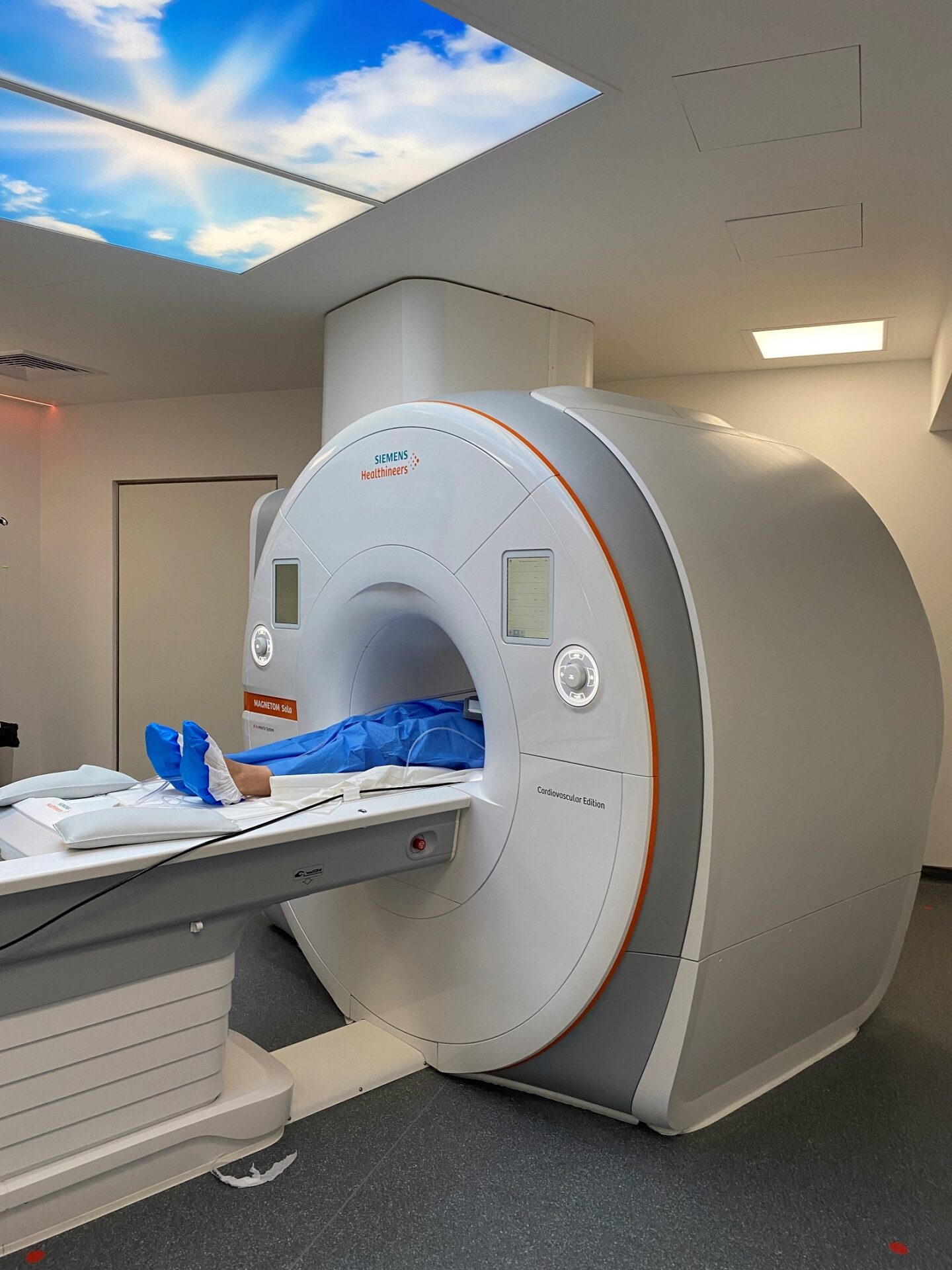
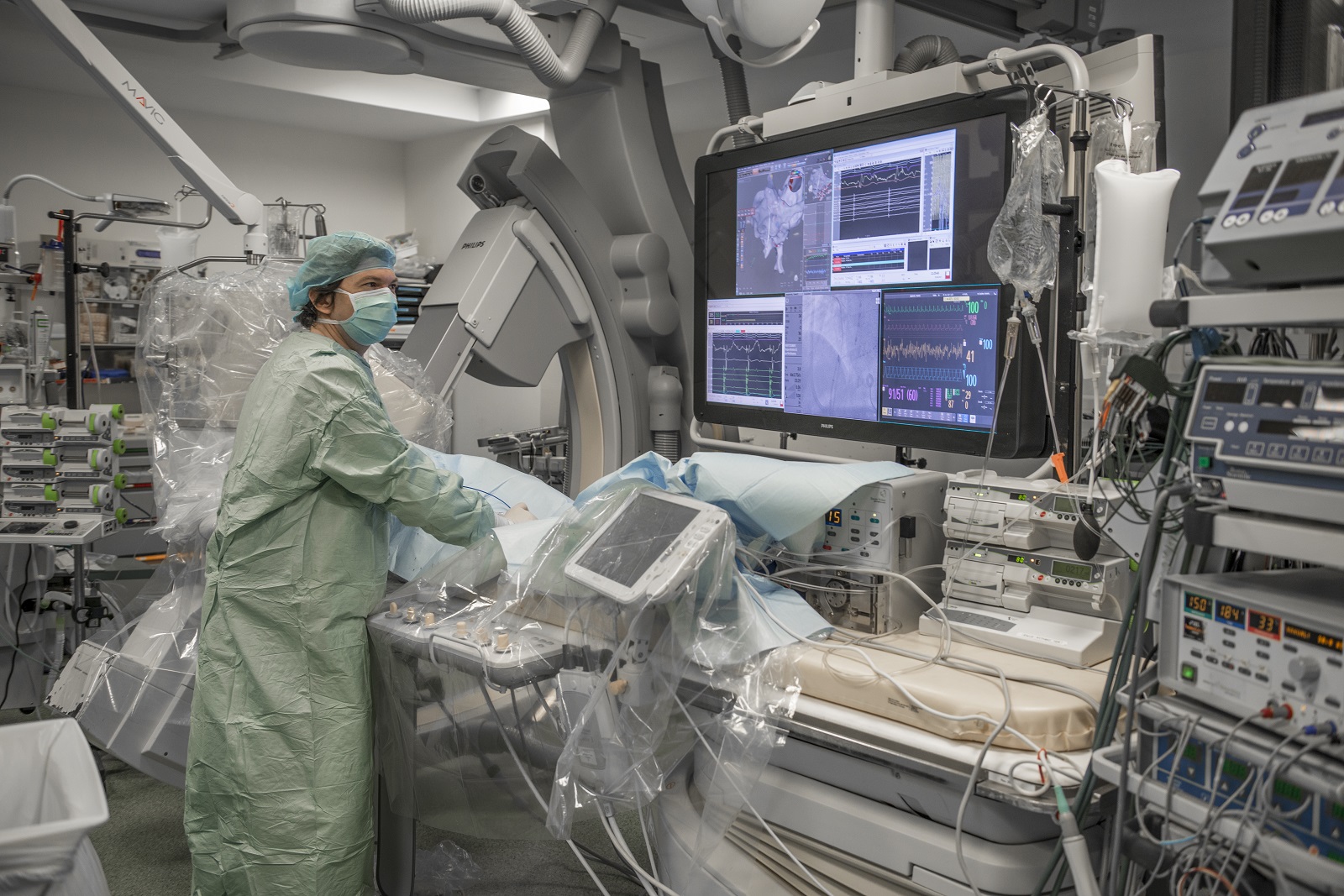
Collaborer avec les équipes de l’ICAN
Nous proposons des expertises à 360° en cardiométabolisme pour accompagner les projets académiques ou industriels : aide au montage de projets, plateau d’investigation clinique, plateformes scientifiques, accompagnement juridique et valorisation de la recherche
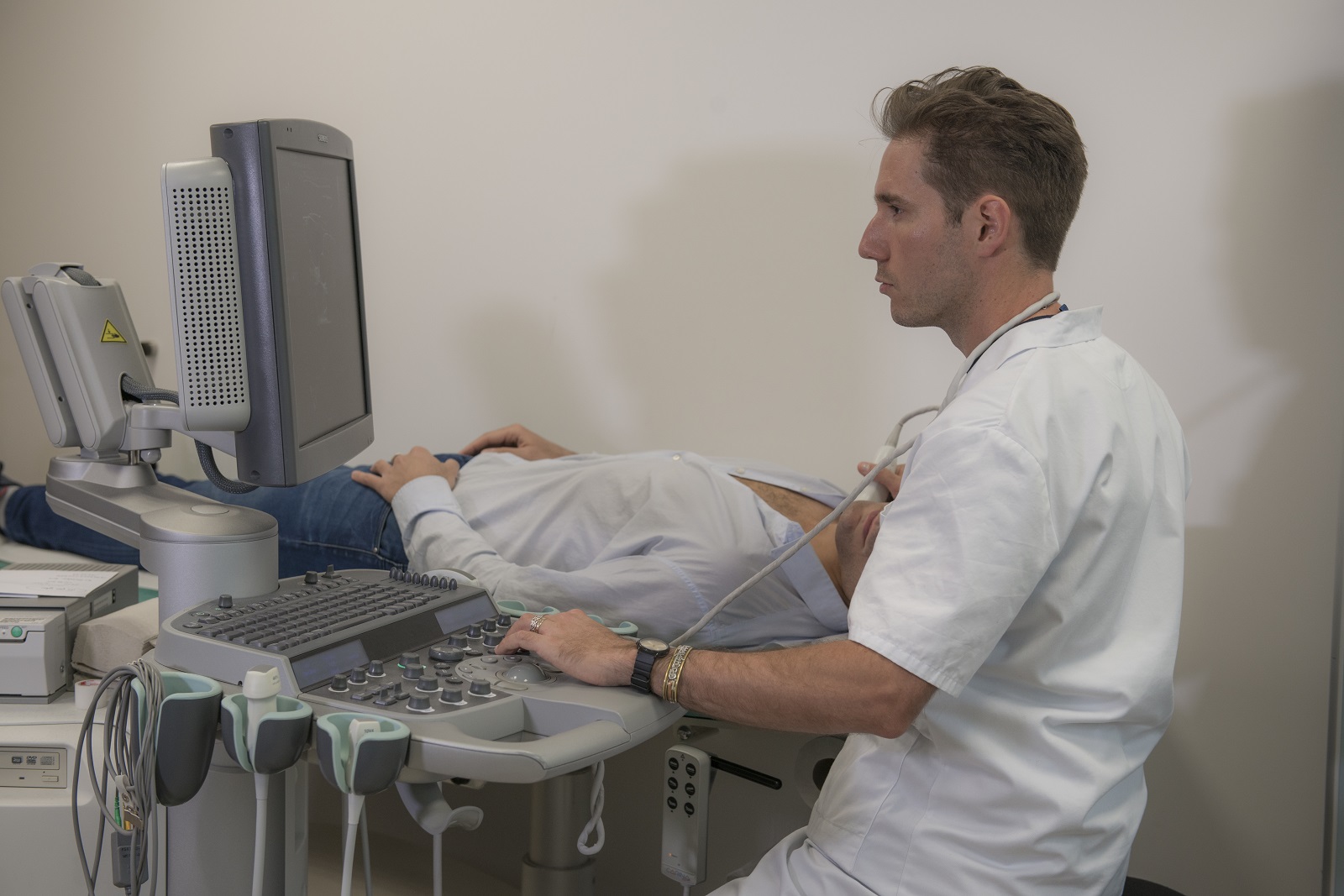
Nos plateaux de recherche clinique
ICAN Clinical Investigation est un plateau d’investigation clinique dédié à la recherche biomédicale sur les maladies du cardiométabolisme. Les équipes d’ICAN Clinical Investigation accompagnent les promoteurs académiques ou industriels dans le montage de leurs essais cliniques.
ICAN Imaging conjugue 3 activités complémentaires : l’acquisition des images avec une IRM cardiovasculaire 1.5T de dernière génération, l’analyse standardisée de référence et la gestion des images par le laboratoire central d’imagerie (Core Lab), ainsi que le montage de projets incluant les aspects méthodologiques, réglementaires, financiers et de communication par l’IHU ICAN.

Nos 6 plateformes scientifiques
L’IHU-ICAN a structuré, sous la forme de plateformes scientifiques, une offre de service unique pour accélérer la recherche sur les maladies du cardiométabolisme. Ces plateformes scientifiques soutiennent la mission de recherche de la communauté scientifique et médicale de l’IHU et elles sont ouvertes aux collaborations académiques et industrielles. L’innovation est au coeur de leur fonctionnement et leur complémentarité est un facteur d’accélération des projets scientifiques.

Situé au cœur de l’hôpital de la Pitié Salpêtrière et s’appuyant sur une communauté scientifique et clinique d’envergure internationale, l’IHU ICAN a noué de nombreux partenariats avec des entreprises pharmaceutiques, des grands groupes aux jeunes sociétés de biotechnologie, dans le cadre d’activités de recherche et de développement.
Soutenez la fondation ICAN
Avec le soutien des donateurs et mécènes, l’IHU ICAN accélère les découvertes au bénéfice des patients sur les maladies du métabolisme.
En soutenant la fondation l’ICAN vous participez au développement d’une prise en charge globale et sur-mesure des maladies du cardiométabolisme. Avec le soutien des donateurs et mécènes, la fondation ICAN accélère les découvertes au bénéfice des patients sur les maladies liées au métabolisme : diabète, obésité, maladie du foie (NASH), maladies cardiovasculaires et invente la médecine de demain dans le cardiométamolisme. Cette médecine dite des 5P : Personnalisée, Participative, Prédictive, Préventive et de Preuve est au cœur de la mobilisation des experts de l’ICAN. Avec votre soutien ils gagneront un temps précieux pour faire progresser la recherche.
Chaque don compte. Merci de votre soutien.


